Science
Related: About this forumA Round Robin Evaluation of the Radiation Stability of Actinide Extraction Reagents for Nuclear Fuel Recovery.
The paper I'll discuss in this post is this one: Interinstitutional Study of the New EURO-GANEX Process Resistance by Gamma Irradiation Test Loops Ivan Sanchez-Garcia, Xavier Heres, Dean. R. Peterman, Hitos Galan, Sylvain Costenoble, Sylvain Broussard, Johann Sinot, Travis S. Grimes, Kash Reid Anderson, Santa Jansone-Popova, Maria Chiara Gullo, Alessandro Casnati, Andreas Wilden, and Andreas Geist Industrial & Engineering Chemistry Research 2023 62 (47), 20326-20339.
Recently in this space, I discussed in this space the ability of a biological protein, lanmodulin, to complex the higher anthropogenic actinide elements americium and curium:
A Bacterial Protein Can Transport Heavy Highly Radioactive Actinides.
From the text of the paper discussed therein, I quoted this excerpt:
Note: I have substituted "approx." for the traditional symbol in the original text, which does not yet show up in DU4.
As I favor the continuous recycling of used nuclear fuels to recover the valuable resources therein, I hope that we will see more - a lot more - use of mixed uranium-plutonium fuels, and thus larger quantities per ton of americium and curium.
The heaviest actinide nuclide that does not have a critical mass occurs naturally, 238U, which makes up the bulk of the billion ton quantities of uranium naturally occurring in the Earth's crust and oceans, even without respect to the planetary mantle, which does cycle through the crust. 238U can be and is transmuted into heavier actinides all of which do have critical masses, including all of the isotopes of neptunium, plutonium, americium and curium, all of which can be isolated from used nuclear fuels, and thus can be utilized to produce clean primary energy.
I have referred to the critical masses of neptunium here: Experimental Determination of the Bare Sphere Critical Mass of Neptunium-237. (Unfortunately the graphics in that post have expired, and I will not have time to restore them.)
I have referred to the critical masses of americium isotopes here:
Critical Masses of the Three Accessible Americium Isotopes. (The graphics in this post are intact.)
Since both elements have critical masses, they are potential sources of clean primary energy. Nonetheless, they are still thought of as "nuclear waste" with the unfortunate use of this evocative but rather stupid and misleading arbitrary locution rather resistant to meaningful address, and thus discussed in terms of their radiotoxicity and heat loads.
Before proceeding further, let me state that I am not a solvent extractant kind of guy, even though almost all of the isolation of plutonium to make MOX fuels (chiefly in France and to a lesser extent, Britain) has relied on an industrial scale solvent extraction process, PUREX (Plutonium/Uranium Reductive EXtraction). The process discussed in this paper is called GANEX (Group ActiNide EXtraction). Both of these involve extraction into water immiscible solvents using complexing agents. In PUREX this solvent has been historically, on an industrial scale, kerosene. Dodecane, another product of dangerous fossil fuels is often considered as an extraction solvent, although in theory this can be made by the deoxygenation of the fatty acid lauric acid from biological matrices.
As opposed to a solvent extraction kind of guy, I am a pyroprocessing kind of guy, in favor of using molten salt chemistry and related chemistry, and or processes known as fluoride volatility, voloxidation, electrorefining and other techniques to separate the 45 or so elements in the periodic table formed in nuclear fuels during fission based operations. These processes require heat, something that fresh used nuclear fuel generates quite readily, or in the case of older used nuclear fuels, can be generated by operating nuclear reactors.
This said, I can see limited applications in which solvent extraction approaches may have limited use, particularly, if desired, the separation of americium and curium from their lanthanide congeners and other lanthanides.
It is the radioactive lanthanides in the presence of actinides that are the focus of the paper under discussion.
One of the many advantages of radiation as a tool is its ability to break chemical bonds. Photosynthesis, for example, utilizes visible light radiation to break water's chemical bonds. Some bonds require more energy to break than those in water do. This is particularly important for the class of ubitiquous pollutants known as "PFAS" which contain multiple carbon fluorine bonds, among the strongest chemical bonds known. The energy to cleave them lies not in the visible spectrum, but in the far UV spectrum; the energy required must have wavelengths on the order of 220 nm or shorter to break these bonds, going into the x-ray and gamma spectrum. When I think of ways to address the very serious issue of PFAS contamination, the use of radioactive fission fragments is very high on my intellectual radar.
In aged used nuclear fuel, some of the lanthanides are no longer highly radioactive: Lanthanum, praseodymium, neodymium and gadolinium can be isolated from these fuels with radioactivity levels consistent with or even less (in the cases of lanthanum and gadolinium) radioactive than the lanthanides found in natural ores. Cerium in fresh used nuclear fuels has a relatively short lived radioisotope 144Ce, with a half life of 284 days, that will require about a decade of cooling for the bulk cerium to consist of stable or metastable isotopes. (With respect to metastability, some natural isotopes of cerium are thought to be slightly radioactive, but with half-lives comparable to the age of the universe.) Europium, and samarium, by contrast, have radioisotopes found in used nuclear fuel that have half-lives on the order of years and decades. In theory, although probably not in practice, both europium and samarium, which have characterized +2 oxidation states, could be isolated by redox procedures, but this would be messy. In nuclear fuels they are almost certainly in their more stable +3 oxidation state and are probably better addressed as such. Promethium does not exist on Earth except, by suspicion but not - to my knowledge - direct observation, in vanishingly small amounts uranium ores from spontaneous fission. All of its isotopes are radioactive, although only one is likely to be in used nuclear fuels 147Pm, with a half-life of 2.62 years. Happily the radioactive lanthanide isotopes of samarium, europium and promethium all have fairly high neutron capture cross sections - they could be used as control rods - and their concentrations are thus suppressed, but they do accumulate and are found in used nuclear fuels. The accumulation of samarium isotopes is responsible for the shut down of used nuclear fuel rods before the fissionable isotopes are consumed.
Solvent extraction relies on the use of molecular complexants, these are organic molecules, and as such, in radiation fields are subject to chemical bond cleavage. That is the focus of this paper on the GANEX process, a concern being the lanthanides that are radioactive.. Note, as well, that all of the actinides are radioactive, in particular curium and americium.
From the introductory text of the paper:
Within the homogeneous strategy, which addresses the recycling of U and the transuranic elements (TRU = Np, Pu, Am, Cm) contained within a single fuel type and distributed homogeneously throughout the reactor core, GANEX (Group ActiNide EXtraction) is the most promising process to recover all of them. (9) In the GANEX concept, bulk uranium is removed in a first cycle, followed by the coextraction of all actinides in a second cycle. Three options exist for this second cycle, the CEA-GANEX, EURO-GANEX, and CHALMEX processes. (4,8) The EURO-GANEX is one of the most promising options to achieve the desired goals (10) and is based on the coextraction of all actinides using a mixture of N,N,N′,N′-tetraoctyl-diglycolamide (TODGA) (11,12) and N,N′-dimethyl-N,N′-dioctyl-2-hexylethoxy-malonamide (DMDOHEMA) (13) extractants. TODGA exhibits high affinity for actinides and lanthanides, but the addition of DMDOHEMA is essential to avoid precipitation caused by the high Pu concentration in the organic phase. (10,14) After the coextraction of An and Ln into the organic phase, a separation between both can be obtained by selective stripping of the actinides as a group, using a mixture of 2,6-bis(5,6-di(sulfophenyl)-1,2,4-triazin-3-yl)-pyridine (SO3-Ph-BTP) (15) and acetohydroxamic acid (AHA). (16) The EURO-GANEX process was tested in an irradiation loop (17) and successfully demonstrated with high Pu content using genuine nuclear fuel solution in centrifugal contactors for the first time, (18) obtaining excellent results. However, EURO-GANEX also has some drawbacks: One of them is that the combination of two extractants in the organic phase (TODGA and DMDOHEMA) complicates solvent management; another one is that the sulfonated BTP reagent employed in the aqueous phase does not accomplish the “CHON principle”, (19) leading to troublesome sulfur-containing byproducts. Therefore, the process needs to be further optimized to meet the criteria mentioned above and to simplify the process as much as possible...
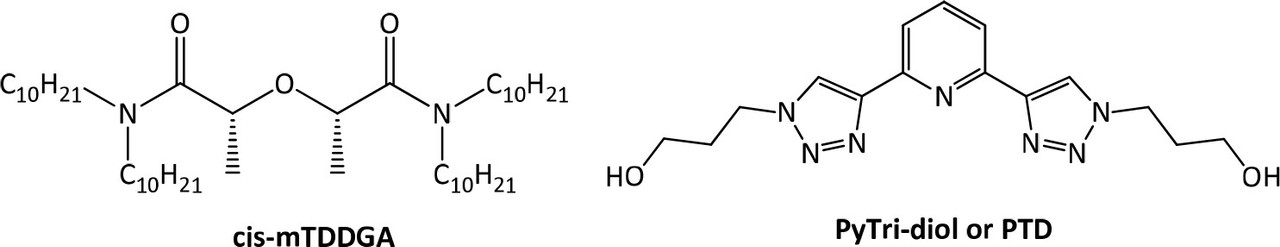
...In order to replace SO3-Ph-BTP, a novel molecule which meets the CHON principle, 2,6-bis[1-(propan-1-ol)-1,2,3-triazol-4-yl)]pyridine (PyTri-Diol, or PTD) is proposed (Figure 1, right). This agent was found to have high actinide selectivity and radiochemical stability (26−31) and could be suitable for the application as a stripping agent not only in GANEX process but also in i-SANEX (Innovative Selective Actinide Extraction) process involved in the heterogeneous strategy of actinides recycling. (28,31,32)
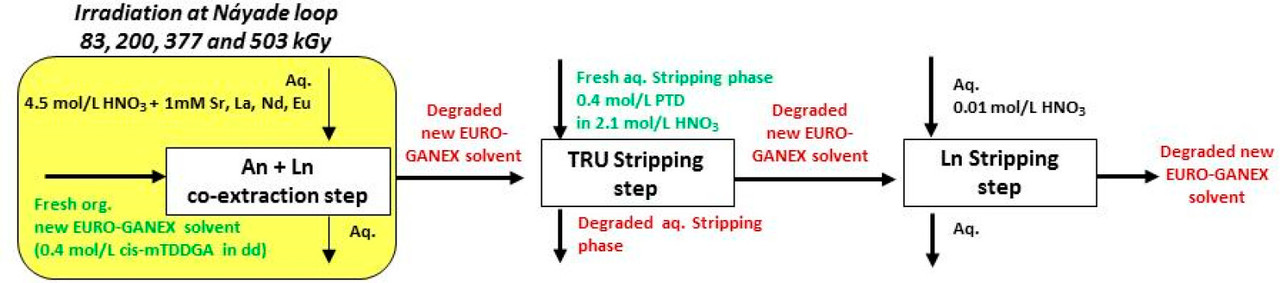
These improvements made to the EURO-GANEX system have resulted in the emergence of the so-called New EURO-GANEX process, with cis-mTDDGA/dodecane (dd) in the organic phase and PTD in the selective TRU back-extraction aqueous solution (Figure 1). As in the EURO-GANEX process, the cis-mTDDGA solvent is useful to extract actinides and lanthanides from an acidic GANEX first cycle raffinate. After this coextraction step, the TRU elements are selectively stripped into the PTD aqueous solution, while cis-mTDDGA maintained rare earth elements in the organic phase...
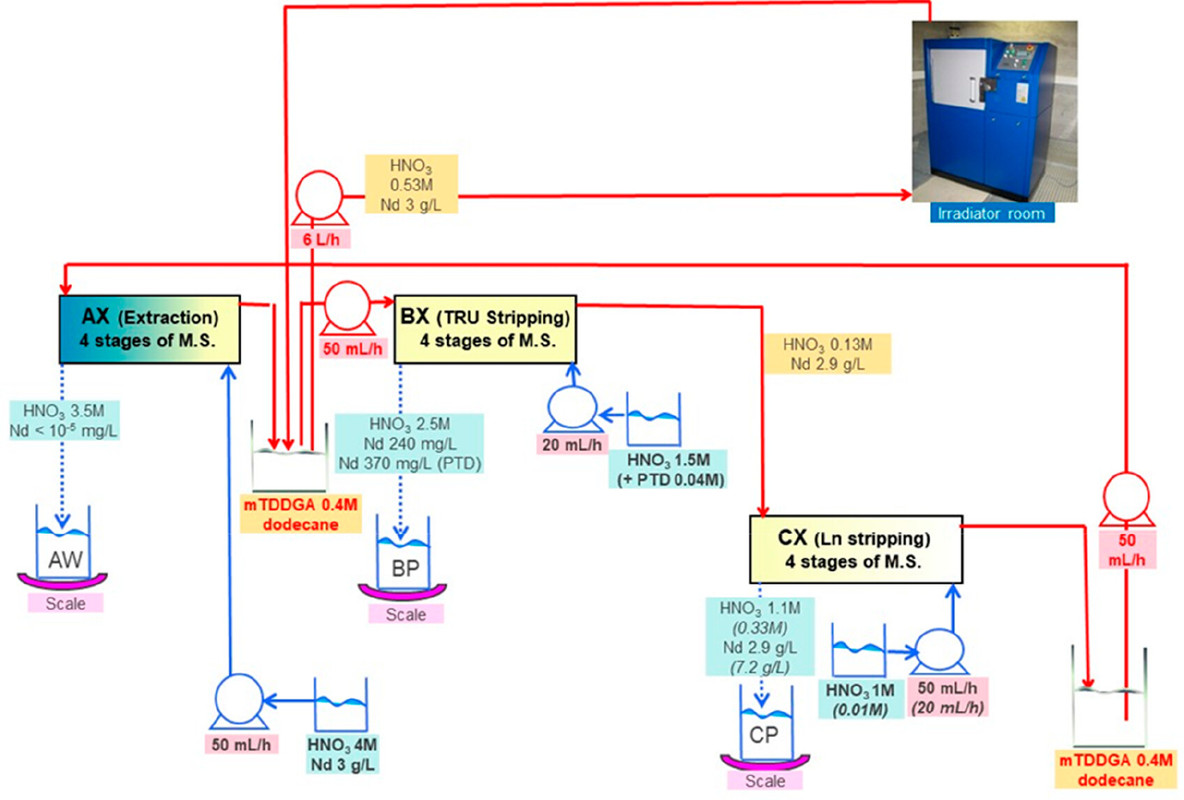
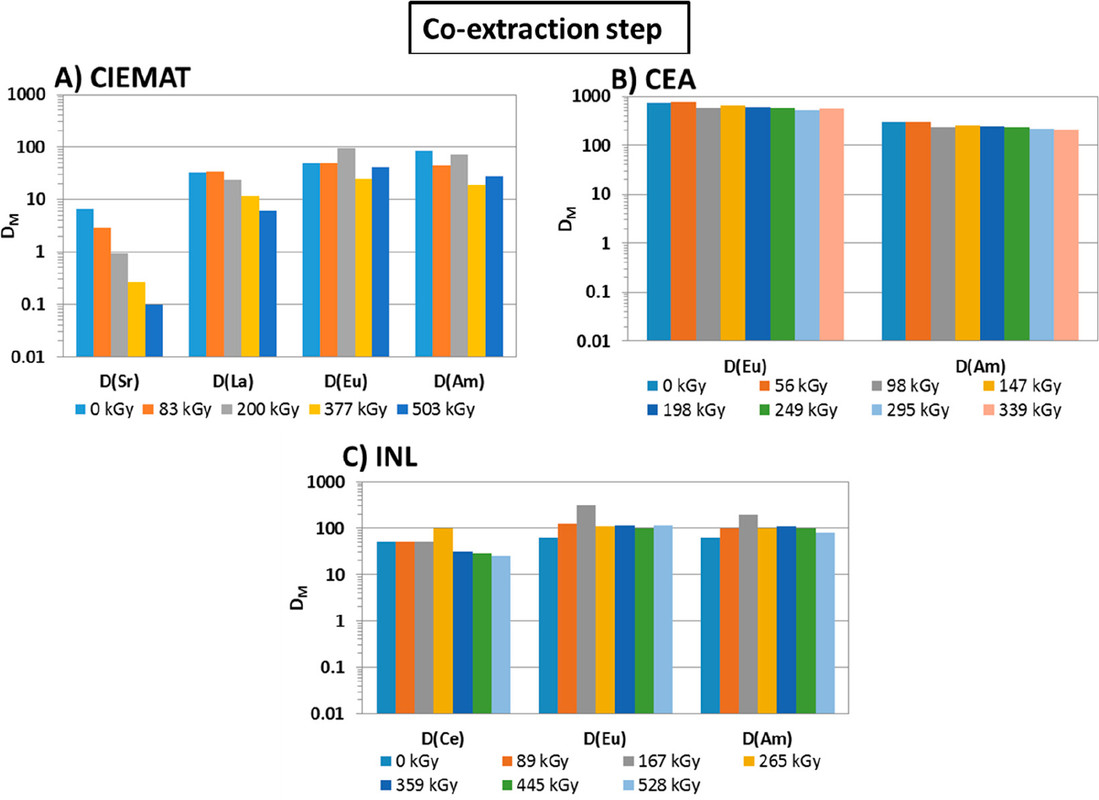
...this work is involved as part of an interinstitutional collaboration between CEA (Commissariat à l’Énergie Atomique et aux Énergies Alternatives) in France, INL (Idaho National Laboratory) in the United States, and CIEMAT (Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas) in Spain. For this collaboration, each research center employed its irradiation loop device to evaluate the resistance of the New EURO-GANEX process to gamma radiolytic degradation using similar conditions and same solutions...
The solvents in this system were subject to radiation exposure using real nuclear fuel solutions as well as radiation generating loops.
Figure 1 from the text:
The caption:
The caption:
Fig
The caption:
Figure 3. Flowsheet of the loop test following the CX hydrodynamic problem (italics indicate initial value). The CX outflow solvent samples for batch studies were taken after 56, 98, 147, 198, 249, 295, and 339 kGy (M.S. = mixer-settler).

The caption:
The caption:
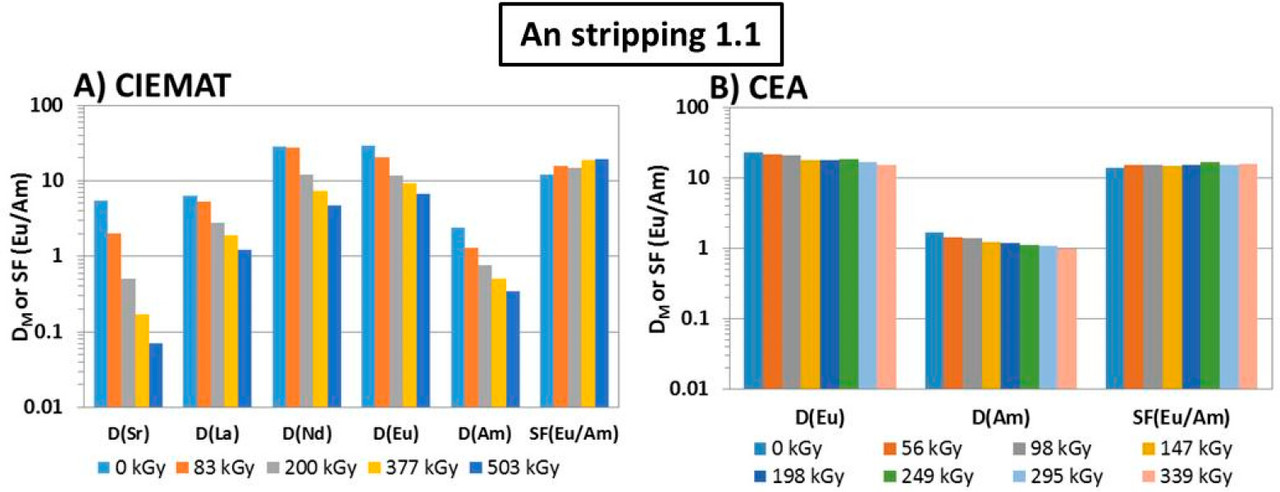
The caption:
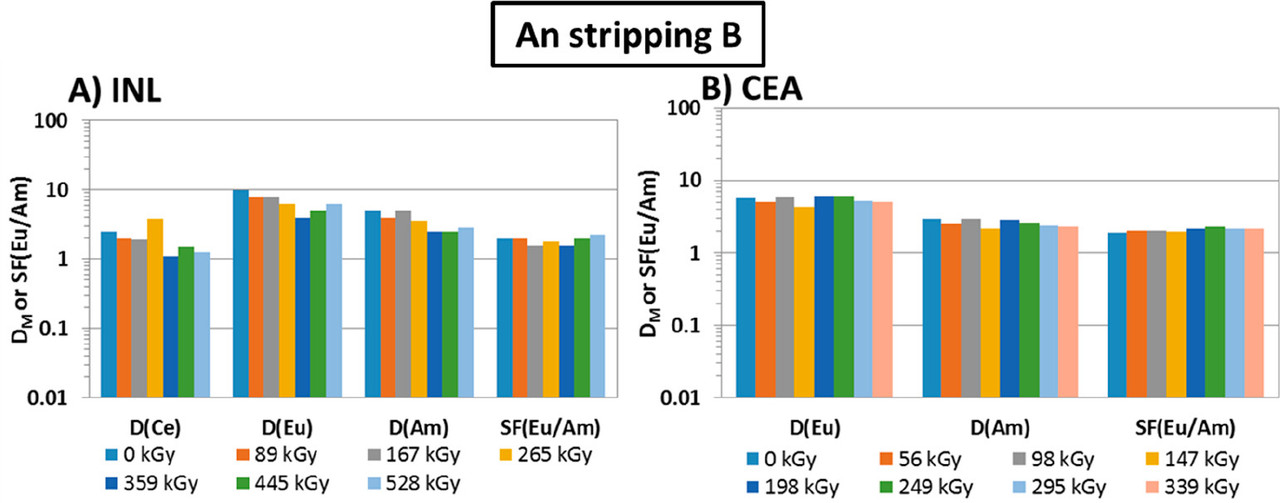
The caption:
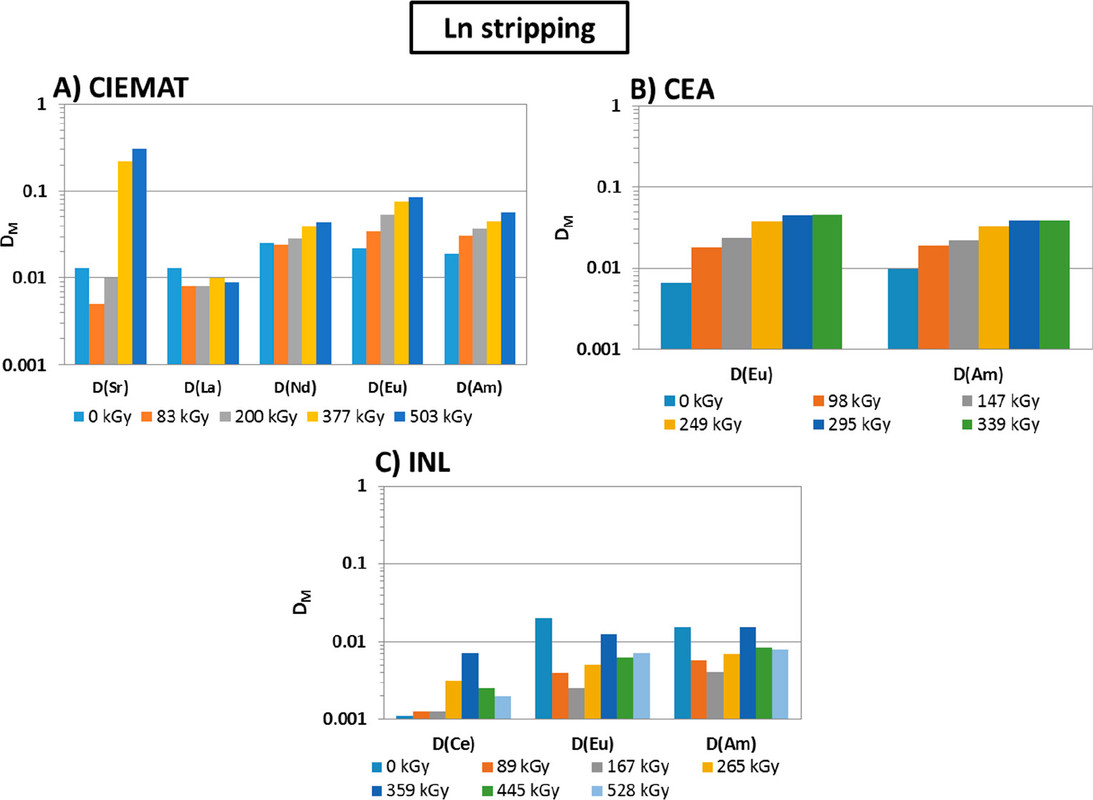
The caption:
The full paper gives structures of some of the degradants found.
The international team of authors discuss, in their conclusions, some of the issues found in the GANEX systems evaluated, and note, with minor differences, the similarity of their results.
From my personal perspective, the situation might be improved by removing neptunium and plutonium (along with uranium) as volatile fluorides before undertaking a GANEX procedure to isolate americium and curium, but nobody cares what I think.
I trust you'll have enjoyable holidays.